
El 'sorteo genético': ¿por qué el hantavirus es más letal para algunas personas que para otras?
Las explicación está en los factores genéticos que controlan la respuesta inmunitaria: la inmunogenética
Publicado el 15/05/2026 a las 09:33
Cuando hablamos de infecciones virales, solemos culpar al patógeno. Sin embargo, en el caso de los hantavirus, virus transmitidos por roedores que pueden causar síndromes cardiopulmonares o renales letales, la gravedad de la enfermedad es, en gran medida, una cuestión de “lotería genética”.
¿Por qué algunas personas solo presentan una infección más leve mientras que otras terminan en cuidados intensivos con una fuga masiva de líquidos en sus pulmones? La respuesta se esconde en nuestro propio ADN; en concreto, en los factores genéticos que controlan la respuesta inmunitaria: la inmunogenética.
LA 'LOTERÍA' DE LOS ALELOS Y LA TORMENTA DE CITOQUINAS
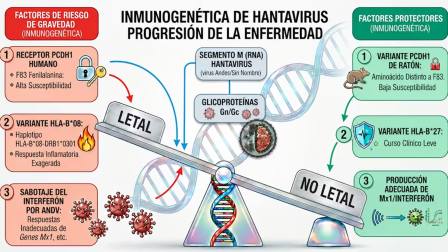
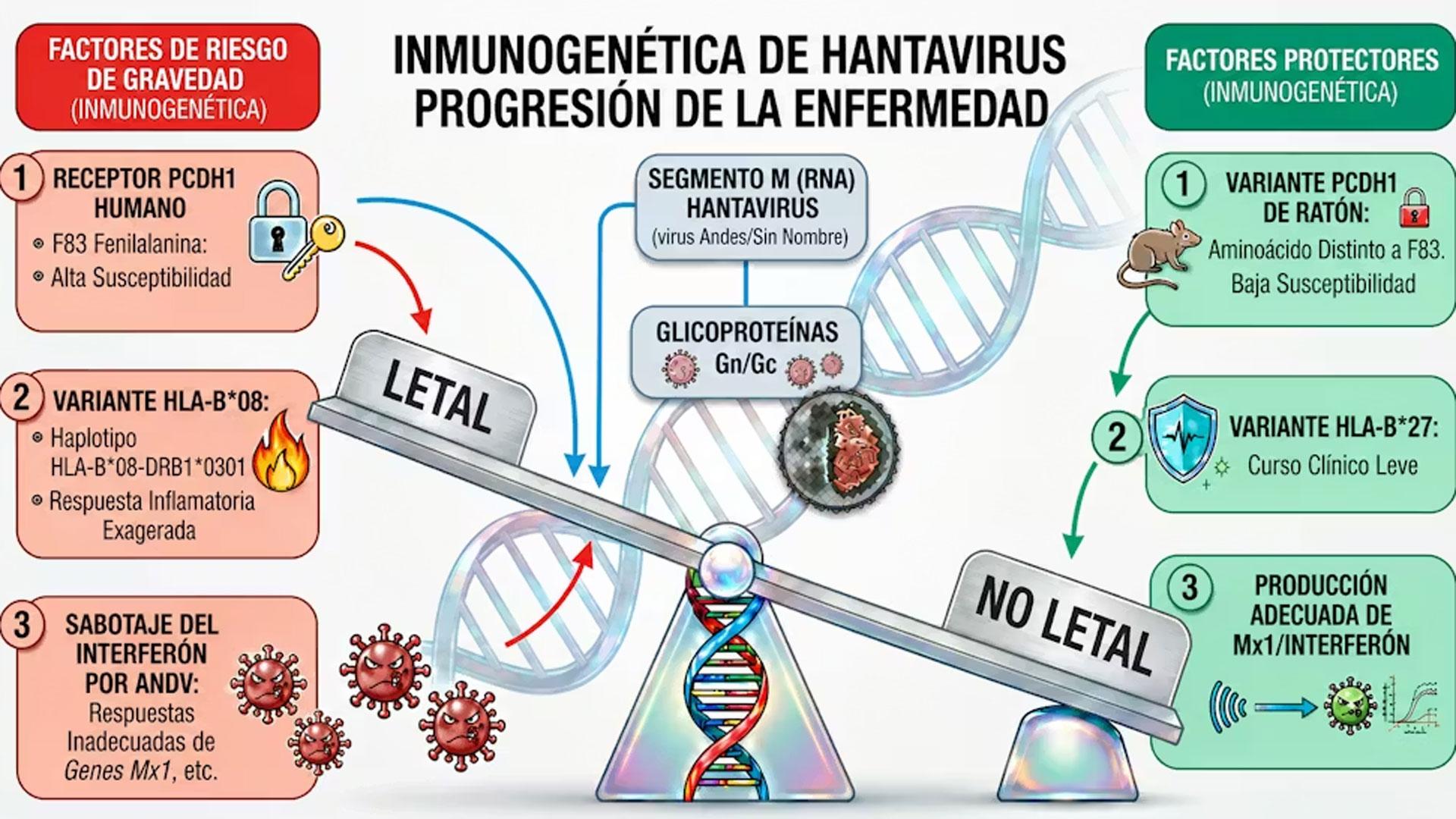
Uno de los grandes protagonistas en cómo reaccionamos frente a una infección es el sistema HLA (siglas en inglés de antígenos leucocitarios humanos), que se encarga de mostrarle a nuestras células inmunitarias fragmentos seleccionados del virus. Sin embargo, no todos los seres humanos presentamos el mismo sistema: existen muchas variantes genéticas (alelos) diferentes de estos genes HLA, y algunas parecen influir en la gravedad de la enfermedad.
Se conoce que ciertas de esas variantes, como el alelo HLA-B*08, actúan como un “empujoncito” hacia una enfermedad grave tras la infección con el hantavirus Puumala (común en el norte de Europa y de Rusia), provocando una respuesta inflamatoria exagerada que daña nuestros propios tejidos. Por el contrario, los afortunados portadores del alelo HLA-B*27 tienden a experimentar un curso clínico mucho más leve y controlado, aunque la interpretación de este tipo de estudios no es tan simple.
Pero antes de que el sistema HLA entre en acción, se libra una batalla silenciosa en nuestras células que conlleva la producción de interferones, proteínas defensivas que son bloqueadas por el hantavirus Andes, el responsable del brote en el crucero MV Hondius. Si, además de este sabotaje, la persona infectada posee variaciones menos adecuadas (producción elevada o disminuida) en los genes que deben responder a esta alarma (como el gen Mx1, encargado de fabricar una proteína clave para frenar la replicación viral), el patógeno gana una ventaja devastadora desde el primer minuto.
Cuando el sistema inmunitario reacciona ante la invasión masiva, entra en pánico y se desata la denominada “tormenta de citoquinas”, en la que se liberan grandes cantidades sustancias inflamatorias sin control. Pequeñas variaciones en el código genético de los genes que codifican dichas citoquinas, sus receptores o mediadores –como el factor de necrosis tumoral (TNF)– determinan si esta inflamación será benéfica o destructiva.
En los pacientes más graves, esta hiperactivación provoca que las paredes de los vasos sanguíneos pierdan su impermeabilidad, causando las temibles hemorragias y la “fuga capilar” que inunda los pulmones o colapsa los riñones.
ATENCIÓN TAMBIÉN A LAS MUTACIONES GENÉTICAS EN EL VIRUS
Aunque no solo las mutaciones de nuestros propios genes influyen en el transcurso de la enfermedad: también lo van a hacer las del propio virus. Antes, hay que recordar cómo se produce la infección.
El receptor principal para las cepas de hantavirus más letales del continente americano, como los virus Andes y Sin Nombre, es una proteína humana llamada protocadherina-1 (PCDH1), que funciona en el control de las uniones entre las células. Este receptor recubre los vasos sanguíneos de nuestros pulmones y también se expresa en el cerebro, los riñones y otros órganos.
En la envoltura externa del virus se encuentran unas pequeñas “espículas”, llamadas glicoproteínas, que se acoplan directamente a ese receptor como si de una llave en una cerradura se tratase.
El ARN del hantavirus está dividido en tres fragmentos –grande (L), mediano (M) y pequeño (S)– y cada uno contiene información para distintas funciones. El “segmento M”, el mediano, es especialmente importante porque codifica para esas glicoproteínas responsables de la entrada viral. Por lo tanto, mutaciones relevantes en el “segmento M” podrían facilitar o dificultar la entrada del virus.
Se sabe que en los humanos hay un aminoácido –fenilalanina F83– en la proteína PCDH1 que es esencial para la entrada del virus. El ratón colilargo, principal sospechoso de hospedar al virus, presenta el mismo aminoácido, mientras que el del ratón común es distinto y por ello no es infectado.
Es decir, el ser humano se ha convertido “por casualidad” en un organismo “hospedador”, con la diferencia de que nuestra respuesta inmunitaria no está tan adaptada al hantavirus como la del ratón.
Y por si fuera poco, el gen PCDH1 posee ciertas variantes que favorecen la hiperreactividad de las vías respiratorias, debido a que la proteína del mismo nombre que produce regula la función de la barrera epitelial de las vías respiratorias a través de mecanismos que aún no están claros. Dichas variantes genéticas podrían tener, pues, un efecto negativo en la infección del hantavirus, aunque no se ha estudiado.
LOS ROEDORES Y SU SISTEMA GENÉTICO ADAPTADO PARA SOBREVIVIR
Curiosamente, los verdaderos maestros en la gestión de este virus son los roedores que lo transmiten. A lo largo de miles de años de evolución conjunta, ratones (y topillos) han ajustado su genética para no enfermar.
Algunos estudios revelan que variaciones en los propios genes de inmunidad innata de los roedores hacen que funcionen con una intensidad diferente. Es el caso del gen Mx2 –el equivalente a nuestro Mx1, citado más arriba–, que produce una proteína (MxA) que evita la replicación del ARN viral; o los receptores TLR4, que reconocen proteínas del patógeno. Estas proteínas con variantes diferentes a las humanas permiten al virus y al animal hospedador llegar a un “acuerdo de paz”. Es decir, el sistema inmunitario del roedor mantiene a raya al virus lo justo para no morir, tolerándolo.
En conclusión, sobrevivir a un hantavirus no depende únicamente de la agresividad del intruso, sino de la arquitectura genética del hospedador que lo recibe. Comprender este complejo equilibrio nos acerca cada vez más a proteger mejor a las personas más vulnerables.
Este artículo fue publicado previamente por la Oficina de Transferencia de Resultados de Investigación (OTRI) de la Universidad Complutense de Madrid (UCM).
Narcisa Martínez Quiles, Catedrática de Inmunología (UCM) y Especialista en Inmunología (Ministerio de Sanidad), Universidad Complutense de Madrid
Este artículo fue publicado originalmente en The Conversation. Lea el original.
